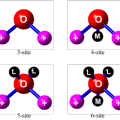
水分子力场种类划分与其它任何分子相比,水分子具有无与伦比的重要性。三力点水分子模型的分子间相互作用力,由静电相互作用和van der Waals相互作用两个部分组成。van der Waals相互作用利用球形对称的Lennard-Jones势函数近似,作用中心位于氧原子上。在分子力场中计入极化效应的分子力场,被称为可极化分子力场。
在MaterialStudio中御用,参数是加密不公开的,虽然lammps也能用,但是参数不全。UFF=Universal Force Field:涵盖整个周期表的普适型力场。大分子力场CHARMM=Chemistry at HARvard Macromolecular Mechanics,函数形式简单,包含以下版本:CHARMM19:联合原子力场,也适用于蛋白质。名字编号是最初使用这种力场的CHARMM版本得名的。这是目前最好的amber力场。