gmx sorient [-f [<.xtc/.trr/...>]] [-s [<.tpr/.tpb/...>]] [-n [<.ndx>]]
[-o [<.xvg>]] [-no [<.xvg>]] [-ro [<.xvg>]] [-co [<.xvg>]]
[-rc [<.xvg>]] [-nice ] [-b ] [-e ] [-dt ]
[-[no]w] [-xvg ] [-[no]com] [-[no]v23] [-rmin ]
[-rmax ] [-cbin ] [-rbin ] [-[no]pbc]
gmx sorient用于分析溶质分子周围的溶剂分子的取向. 它可以计算从一个或多个参考位置到每个溶剂分子第一个原子的向量(向量 A⃗ )与另外两个向量之间的角度:
θ1: 向量 A⃗ 与从溶剂分子第一个原子到第二和第三个原子中点的向量之间的夹角
θ2: 向量 A⃗ 与由三个原子定义的溶剂分子平面的法线之间的夹角, 或者, 当使用-v23选项时, 向量 A⃗ 与从原子2到原子3的向量之间的夹角.
参考位置可以是一组原子或是一组原子的质心. 溶剂原子组中的每个溶剂分子只能包含3个原子. 对每一帧, -o和-no选项只会考虑处于-rmin和-rmax之间的溶剂分子.
-o: rmin≤r≤rmax 范围内 cosθ1 的分布
-no: rmin≤r≤rmax 范围内 cosθ2 的分布
-ro: ⟨cosθ1⟩ 和 ⟨3cos2θ2−1⟩ 与距离 r 的函数关系
-co: 对距离 r 范围内所有溶剂分子的 cosθ1 和 3cos2θ2−1 进行加和, 得到它们与 r 的函数关系
-rc: 溶剂分子的分布与 r 的函数关系
输入/输出文件选项| 选项 | 默认值 | 类型 | 说明 |
|---|
-f [<.xtc/.trr/...>] | traj.xtc | 输入 | 轨迹: xtc trr cpt trj gro g96 pdb tng |
-s [<.tpr/.tpb/...>] | topol.tpr | 输入 | 结构+质量(db): tpr tpb tpa gro g96 pdb brk ent |
-n [<.ndx>] | index.ndx | 输入, 可选 | 索引文件 |
-o [<.xvg>] | sori.xvg | 输出 | xvgr/xmgr文件 |
-no [<.xvg>] | snor.xvg | 输出 | xvgr/xmgr文件 |
-ro [<.xvg>] | sord.xvg | 输出 | xvgr/xmgr文件 |
-co [<.xvg>] | scum.xvg | 输出 | xvgr/xmgr文件 |
-rc [<.xvg>] | scount.xvg | 输出 | xvgr/xmgr文件 |
控制选项| 选项 | 默认值 | 说明 |
|---|
-nice <int> | 19 | 设置优先级 |
-b <time> | 0 | 从轨迹文件中读取的第一帧(ps) |
-e <time> | 0 | 从轨迹文件中读取的最后一帧(ps) |
-dt <time> | 0 | 只使用t除以dt的余数等于第一帧时间(ps)的帧, 即两帧之间的时间间隔 |
-[no]w | no | 查看输出的.xvg, .xpm, .eps和.pdb文件 |
-xvg <enum> | xmgrace | xvg绘图格式: xmgrace, xmgr, none |
-[no]com | no | 使用质心作为参考位置 |
-[no]v23 | no | 使用原子2和3之间的向量 |
-rmin <real> | 0 | 最小距离(nm) |
-rmax <real> | 0.5 | 最大距离(nm) |
-cbin <real> | 0.02 | 余弦的分格宽度 |
-rbin <real> | 0.02 | 距离 r 的分格宽度(nm) |
-[no]pbc | no | 计算质心时检查PBC. 只有当你的参考组包含多个分子时, 才需要使用此选项. |
补充说明
此程序特别适用于计算溶质分子周围水分子的角度分布.
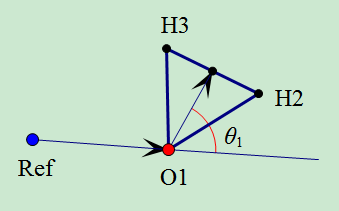
设溶质为单原子离子或分子质心Ref, 溶剂为水分子原子1为O, 原子2和3为H, 则 θ1 对应Ref至O的向量 A⃗ =R⃗ Ref−R⃗ O 与O至两个H连线中点的向量 R⃗ OH2+R⃗ OH32 之间的夹角, 后一向量的方向与水分子偶极矩的方向相同. 因此, θ1 可视为溶质分子周围水分子偶极矩的取向. θ2 对应 A⃗ 与水分子平面法线的夹角. 当使用-v23选项时, 则为 A⃗ 与两个H连线 R⃗ H3−R⃗ H2 之间的夹角.
文章链接:GROMACS各类程序(名称排序)|Jerkwin
如有侵权联系我,我将删除
本文目的只为宣传使用


